
Hipotalamitis: ¿una nueva enfermedad endocrina autoinmune?
11 de mayo 2021
Una revisión de la literatura y el informe de un caso clínico
Hypothalamitis: A Novel Autoimmune Endocrine Disease. A Literature Review and Case Report
UÄur Türe y Col. The Journal of Clinical Endocrinology & Metabolism, 2021, Vol. 106, No. 2, e415–e429
Resumen:
La relación entre el sistema endocrino y la autoinmunidad ha sido reconocida durante mucho tiempo, y uno de los mejores ejemplos de enfermedad endocrina autoinmune es la hipofisitis autoinmune. Una mejor comprensión de los mecanismos autoinmunitarios y los desarrollos radiológicos, bioquímicos e inmunológicos ha dado lugar a la definición de nuevos trastornos autoinmunitarios, incluidos los trastornos hipotalámicos-hipofisarios. Sin embargo, si la hipotalamitis puede ocurrir como una entidad distinta sigue siendo un tema de debate.
Aquí se describe a una mujer de 35 años con masa supraselar en crecimiento, silla turca parcialmente vacía, diabetes insípida central, hipopituitarismo e hiperprolactinemia.
El examen histopatológico de la masa supraselar extirpada quirúrgicamente reveló un infiltrado linfocítico que sugería una enfermedad autoinmune con afectación hipotalámica. La presencia de anticuerpos antihipotálamo contra las células secretoras de arginina vasopresina (AVPcAb) en títulos elevados y la ausencia de anticuerpos antipituitarios sugirieron el diagnóstico de hipotalamitis aislada. Algunas condiciones similares se han informado a veces en la literatura, pero hasta ahora nunca se ha informado el doble hallazgo simultáneo de infiltrado linfocítico y la presencia de AVPcAb.
Conclusiones: la hipotalamitis puede considerarse una nueva enfermedad autoinmune aislada que afecta al hipotálamo mientras que la infundibuloneurohipofisitis linfocítica puede ser consecuencia de una hipotalamitis con posterior afectación autoinmune de la hipófisis. Hasta donde se conoce, esta es la primera observación de afectación hipotalámica autoinmune con diabetes insípida central, silla turca parcialmente vacía, anticuerpos antihipotalámicos e hipopituitarismo.
Comentario:
La participación del sistema endocrino en el contexto de la tolerancia inmune alterada se reconoció por primera vez hace aproximadamente 95 años. Con el correr de los años se ha demostrado que los órganos endocrinos se ven afectados por daños relacionados con la autoinmunidad, y la glándula pituitaria se convirtió en la cuarta glándula endocrina afectada por la autoinmunidad. El síndrome autoinmune poliendocrino (APS), relacionado con la autoinmunidad, consta de compromiso de diferentes órganos endocrinos y se clasifica en 4 categorías; y la hipofisitis posiblemente participe de las 4.
Hay anticuerpos contra antígenos hipofisarios o hipotalámicos asociados con la deficiencia hipofisaria en pacientes con APS-1. La hipofisitis autoinmune (HA), o hipofisitis linfocítica, es una enfermedad hipofisaria relacionada con la autoinmunidad. Puede ocurrir como una enfermedad aislada que afecta solo a la glándula pituitaria o puede estar asociada con la afectación de otros órganos endocrinos, como enfermedades tiroideas autoinmunes. Según la afectación anatómica de la glándula pituitaria, la AH se describe tradicionalmente como adenohipofisitis linfocítica (LAH), cuando solo se afecta la glándula pituitaria anterior, infundibuloneurohipofisitis linfocítica (LINH), cuando el tallo hipofisario y la glándula pituitaria posterior están involucrados, y la panhipofisitis linfocítica (LINH), en el que está involucrada toda la glándula pituitaria.
Los pacientes con HA pueden presentar distintos hallazgos radiológicos asociados con grados variables de hipopituitarismo y/o hiperprolactinemia y diabetes insípida central (CDI).
Hasta hace poco, la hipotalamitis no se ha reconocido como una entidad distinta, y la afectación hipotalámica que se presentaba como una masa supraselar ha sido interpretada como una continuación ascendente de la HA.
Datos recientes sugieren que la afectación autoinmunitaria del hipotálamo que se presenta con una masa supraselar puede no ser un componente de la HA, sino una enfermedad autoinmunitaria aislada llamada hipotalamitis. En esta revisión, a partir de la descripción de un caso con hallazgos clínicos, de laboratorio y de imagen que sugieren hipotalamitis aislada, y se discute si la hipotalamitis es una entidad clínica distinta o no a la luz de los datos actuales.
Reporte de un caso
Una mujer de 35 años con 2 hijos presentó una historia de 2 años de amenorrea, poliuria (5-10 L/día) -polidipsia (mayor de 5 L/día) y aumento de peso. La historia clínica mostró que la paciente tuvo su último parto cuando tenía 30 años y estuvo lactando y tuvo menstruaciones regulares hasta 2 años antes. En el examen físico el examen del campo visual no reveló ninguna anomalía. La investigación hormonal y bioquímica basal mostró hiperprolactinemia; IGF-1 disminuida, hipotiroidismo central con ATPO neg; hipogonadismo central; hipoadrenalismo central y densidad urinaria de 1,005 (1,015-1,025). La investigación inmunológica reveló la presencia de anticuerpos antihipotalámicos (AHA) en títulos elevados; 1/128 (normal, Ë1/8), dirigido contra células secretoras de arginina vasopresina (AVP). Los anticuerpos antipituitarios (APA) fueron negativos. La resonancia magnética (IRM) mostró una lesión levemente hipointensa en el hipotálamo y con falta del punto brillante de neurohipófisis. La lesión era levemente hiperintensa en las imágenes potenciadas en T2 con un realce heterogéneo en T1 con contraste. Las dimensiones máximas A-P, transversal y craneocaudal de la lesión se midieron como 8 Ã 11 Ã 10 mm, respectivamente.
La paciente fue reevaluada 3 meses después y se detectó una progresión en el tamaño de la lesión con lo que se planificó la resección microneuroquirúrgica.
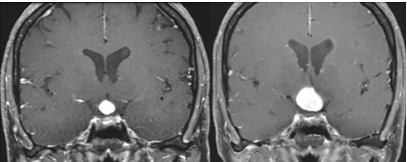
RMN al diagnóstico y a los 3 meses
La paciente fue intervenida y la masa fue extirpada por completo mediante un abordaje pterionaltransilvio (UT) del lado derecho.
La RM cerebral postoperatoria precoz del primer día postoperatorio confirmó la exéresis total de la masa. Durante el postoperatorio se le diagnosticó hipopituitarismo, hiperprolactinemia e ICD.
El examen histopatológico mostró una gran infiltración que consistía predominantemente en linfocitos maduros y células plasmáticas acompañadas de cuerpos de Russell dispersos y pocos histiocitos, sin ninguna evidencia sólida de células germinales atípicas. Se excluyeron los tumores gliales, metastásicos y de células germinales.
La masa extirpada era compatible con un pseudotumor inflamatorio que mostraba características de hipotalamitis linfocítica con abundante infiltración por linfocitos, células plasmáticas y células dendríticas en el tejido resecado, clínicamente acompañado de silla vacía parcial, CDI, hiperprolactinemia, e hipopituitarismo.
Hipotalamitis autoinmune
El término "autoinmunidad hipotalámica-hipofisaria" incluye un amplio espectro de trastornos que incluyen distintas formas de hipofisitis, así como otros trastornos primarios y secundarios endógenos y iatrógenos, caracterizados por infiltración linfocítica de la pituitaria anterior y/o posterior que, algunas veces, afecta al hipotálamo. La hipofisitis autoinmune (HA) es una enfermedad conocida desde hace mucho tiempo y se caracteriza por una infiltración linfocítica de la glándula pituitaria y puede ser primaria o secundaria. El síndrome de Sheehan se define como necrosis hipofisaria posparto y el hipotálamo también puede verse afectado. La hipofisitis anti-PIT-1 es una enfermedad autoinmune descrita recientemente que se caracteriza por deficiencias adquiridas de hormona del crecimiento, prolactina y tirotropina asociadas con autoinmunidad a PIT-1. La meningitis infecciosa puede causar hipopituitarismo y puede estar asociada con la aparición de Ac antipituitarios (APA) y Ac antihipotalámicos (AHA). La autoinmunidad puede ser la causa subyacente del trastorno y afectar el curso de la enfermedad en algunos de los otros trastornos hipotalámicos hipofisarios, incluida la enfermedad relacionada con IgG-4, inhibidores de puntos de control inmunológico, lesión cerebral traumática y tumores cerebrales.
La hipotalamitis autoinmune se describe como una condición autoinmune en la cual el hipotálamo es infiltrado por células inflamatorias que consisten en linfocitos, células plasmáticas, células dendríticas e histiocitos. Es una condición poco común descrita en la literatura actual con muy pocos casos en todo el mundo.
El hipotálamo juega un papel vital en la homeostasis, y su daño predispone a la disfunción hipofisaria, trastornos del comportamiento, alteraciones metabólicas y alteraciones neuroendocrinas. El tallo hipofisario se encuentra entre la hipófisis y el hipotálamo y contiene elementos neurales y vasculares. Durante la embriogénesis, el lóbulo posterior de la glándula pituitaria se desarrolla a partir del infundíbulo, una región específica del diencéfalo ventral y en la glándula madura se compone de terminales axónicos hipotalámicos de neuronas magnocelulares del núcleo paraventricular y del núcleo supraóptico. Entonces, el lóbulo posterior es la continuación del hipotálamo.
Se pueden detectar numerosos anticuerpos en enfermedades variables que involucran el sistema hipotalámico-pituitario. En AH se informaron autoanticuerpos contra la hormona del crecimiento, α-enolasa, secretogranina II, factores específicos de la glándula pituitaria 1a y 2, y se sugirió que estos anticuerpos se producen debido a alteraciones en el tejido pituitario. El papel de la autoinmunidad y el hipotálamo está menos descrita. Una de las principales manifestaciones de la afectación hipotalámica en la autoinmunidad es la ICD. Se detectaron anticuerpos contra las células secretoras de vasopresina del hipotálamo humano en el 36,7% de los pacientes con diabetes insípida idiopática, pero en ninguno de los sujetos de control, lo cual sugiere que la autoinmunidad se extiende al hipotálamo y que los anticuerpos de las células de vasopresina pueden ser marcadores útiles para el diagnóstico de diabetes insípida de origen autoinmune. En los últimos años, los estudios han confirmado nuevos marcadores/antígenos autoinmunes, y anticuerpos que desempeñan un papel en el desarrollo de infundibuloneurohipofisitis linfocítica (LINH). La rabfilina-3A, es una proteína de membrana responsable de la regulación dependiente del calcio de la exocitosis de las vesículas secretoras, y se encuentra en neuronas y células neuroendocrinas. Parecería que los autoanticuerpos contra la rabfilina-3A, pueden usarse como biomarcador en el diagnóstico de LINH. Por otro lado, encontraron autoanticuerpos contra las células secretoras de AVP con ICD. La presencia de autoanticuerpos de células de vasopresina circulantes en pacientes con ICD idiopática puede sugerir una afectación autoinmune del sistema neurohipofisario en el contexto de hallazgos clínicos y radiológicos apropiados.
No se ha discutido suficientemente en la literatura si la HA también puede involucrar al hipotálamo y conducir a hipotalamitis como continuación de la hipofisitis, o si la hipotalamitis es una entidad clínica aislada. Al menos hasta donde conocemos, el primer paciente al que se le diagnosticó una masa supraselar como HA asociada con la silla turca vacía e hipopituitarismo en la literatura se informó hace 20 años. En ese caso, no se detectaron anticuerpos. El examen histopatológico confirmó HA, que pudo haberse extendido al hipotálamo y producir un cuadro clínico caracterizado por masa supraselar, CDI, silla turca vacía e hipopituitarismo.
En caso de afectación autoinmune de la glándula pituitaria y/o el hipotálamo, la glándula pituitaria puede destruirse y volverse más pequeña con los años, por lo que el proceso autoinmune puede resultar en la silla turca vacía como resultado final. La silla turca vacía en pacientes con HA con o sin compromiso hipotalámico puede deberse a una falla de las hormonas hipotalámicas tróficas que conducen a la atrofia del tejido hipofisario y la destrucción autoinmune de la propia glándula pituitaria, respectivamente.
La hipotalamitis también puede ser desencadenada por fármacos inmunomoduladores, y también puede ser una presentación localizada de encefalitis límbica paraneoplásica (PLE) donde los antígenos asociados llevan a la aparición de anticuerpos contra el cáncer acompañante. Los antígenos en determinadas localizaciones del sistema nervioso central como el hipotálamo actúan de forma similar y provocan un síndrome paraneoplásico expresado por pérdida neuronal e inflamación.
Los trastornos alimentarios como la anorexia nerviosa y la bulimia nerviosa pueden estar asociados con el desarrollo de anticuerpos contra la hormona estimulante de los melanocitos α, la adrenocorticotropina y la hormona liberadora de la hormona luteinizante, aunque su papel en la patogénesis de la anorexia nerviosa y la bulimia nerviosa es desconocido.
Anteriormente, no se informó de ningún caso que describiera hipotalamitis secundaria a múltiples endocrinopatías. Por lo tanto, las particularidades microvasculares del hipotálamo podrían ser diferentes de los otros órganos endocrinos y que definirían al hipotálamo como un órgano aislado. El hipotálamo es de origen neuroectodérmico, mientras que los otros órganos endocrinos se originan principalmente en el endodermo. Por lo tanto, no solo su microvasculatura divergente, sino que sus constituyentes gliales y neuronales debido a su origen neural podrían ser la razón subyacente de su rareza, pero esto necesita una mayor aclaración clínica, experimental y molecular.
En la literatura actual, la hipotalamitis autoinmune, ya sea descrita como una entidad secundaria o aislada, se presenta más comúnmente con ICD, amenorrea u oligomenorrea, cambios de comportamiento y alteraciones visuales. En algunos casos también se describieron déficit de memoria, irritabilidad, cefaleas y astenia. La presencia de AHA parece ser una característica distintiva incluso en ausencia de APA en algunos casos. Los APA y AHA podrían ser útiles para confirmar casos sospechosos de trastornos autoinmunitarios hipofisarios-hipotalámicos sobre la base de datos clínicos y radiológicos sugestivos. En estudios previos, la detección de anticuerpos contra las células vasopresina (AVPcAb) en la mayoría de los pacientes con ICD sugirió una afectación autoinmune de esta enfermedad.
Características radiológicas de la hipotalamitis
La hipotalamitis no posee características específicas en la resonancia magnética para distinguirla de otras patologías supraselares como gliomas, metástasis y encefalitis. Hay leve hipo o isointensidad en T1 e hiperintensidad leve en T2 con respecto a la sustancia blanca normal, y estas son características comunes para la mayoría de las patologías hipotalámicas. La falta de brillo de la neurohipófisis en T1 se encontró tanto en el caso actual como en la mayoría de los estudios previos. Aunque el tallo infundibular tiene un grosor normal, la ausencia de una señal T1 alta de la neurohipófisis probablemente se deba a la interrupción entre la neurohipófisis y el hipotálamo. El grosor del tallo hipofisario se encuentra aumentado en algunos estudios previos, pero generalmente sigue siendo de tamaño normal como en el caso actual. El realce heterogéneo de orientación periférica con o sin edema del quiasma óptico o del tracto es un hallazgo común. Una combinación de características de resonancia magnética con la hiperdensidad de la tomografía computarizada es extremadamente útil para el diagnóstico preciso de hipotalamitis. Además de las características inmunohistoquímicas, la resonancia magnética de nuestro caso no mostró indicios de inflamación hipofisaria que se extendiera a la región hipotalámica.
Diagnóstico diferencial de hipotalamitis
El diagnóstico y el diagnóstico diferencial de la hipotalamitis no son fáciles por ser un concepto nuevo reportado en la literatura y por el reducido número de casos. Cuando se examinan los casos considerados hipotalamitis reportados en la literatura y el caso aquí presentado, los 3 motivos más importantes de ingreso son la masa supraselar, la ICD y la insuficiencia pituitaria anterior. Al igual que en AH, la mayoría de los pacientes son mujeres. No hay ningún hallazgo radiológico patognomónico de hipotalamitis, por lo que el examen histopatológico es esencial para el diagnóstico definitivo. Cuando el examen histopatológico muestra que la anomalía subyacente es compatible con la autoinmunidad, el APA y AHA séricos son útiles para la detección de títulos altos de anticuerpos contra células hipotalámicas específicas, lo que hace que el diagnóstico de hipotalamitis sea aún más fuerte. La negatividad de APA también apoya que la lesión es compatible con hipotalamitis.
A la luz de todos estos datos, se proponen criterios mayores y menores para el diagnóstico de hipotalamitis autoinmune (Ver Tabla 1). Cualquier trastorno que afecte el área hipotalámica puede caracterizarse por hipopituitarismo anterior e ICD. Por tanto, en primer lugar conviene desvelar si la lesión es de origen autoinmune o no. Cuando todos los hallazgos (incluido el examen histopatológico de una masa supraselar extirpada quirúrgicamente) revelan afectación autoinmunitaria del hipotálamo, el siguiente paso es aclarar si la masa es hipotalamitis aislada o afectación autoinmunitaria del hipotálamo debido a un trastorno secundario.

Tabla 1: Criterios propuestos para el diagnóstico de hipotalamitis
Tratamiento de la hipotalamitis
No existe suficiente experiencia en el tratamiento de la hipotalamitis. La literatura sugiere glucocorticoides, plasmaféresis y posteriormente inmunoglobulina intravenosa como tratamiento de primera línea; y rituximab, ciclofosfamida y azatioprina como tratamiento de segunda línea. Si bien el tratamiento médico puede resultar en alguna mejora radiológica, no se ha informado ninguna mejora endocrinológica. La cirugía representa el tratamiento más común ya que es el único método que permite el diagnóstico definitivo mediante investigación histológica.
Se puede concluir que se describe un raro caso de hipotalamitis autoinmune aislada luego de eliminar sistemáticamente la posibilidad de su presentación como manifestación secundaria.
Cuando se toman todos los hallazgos mencionados, los datos actuales sugieren que la hipotalamitis puede existir como una enfermedad autoinmune distinta que se presenta con una lesión supraselar, CDI e hipopituitarismo. Sería difícil hacer un diagnóstico diferencial entre la hipotalamitis aislada y la afectación hipotalámica de la HA, pero los marcadores autoinmunes serían útiles. El lóbulo anterior de la hipófisis no está afectado y la ICD es la principal queja en los pacientes con LINH. Por lo tanto, LINH puede ser un componente de la hipotalamitis más que una parte de la HA. Pero no se puede descartar una hipofisitis previa teniendo en cuenta la presencia de silla turca vacía en el paciente, lo que, como se sabe, podría ser un signo indirecto de la involución a menudo autoinmune de la hipófisis. La confirmación histopatológica es el gold standard para el diagnóstico de hipotalamitis autoinmune respaldada por la presencia de AHA. Se requiere una inmunohistoquímica completa, así como APA y AHA que reconozcan las células secretoras de vasopresina para ayudar en el diagnóstico diferencial.
Se necesitan más estudios para aclarar los mecanismos subyacentes responsables del desarrollo de la afectación autoinmune del hipotálamo y si LINH es realmente un subgrupo de AH o es en realidad una extensión de la hipotalamitis. Los autores creen que es necesario revisar la clasificación de la afectación autoinmune del sistema hipotalámico-pituitario.
Copyright 2021. Endoweb.net
Comentarios (-)
Todavía no hay comentarios en este artículo. ¡Nos encantaría conocer tu opinión!